Introduction
Protein-peptide docking and MD rescoring are useful for prioritizing peptide hits before expensive experiments. More specific than small molecules and simpler to design than full proteins or antibodies, peptides have become particularly attractive to biopharma. However, designing a peptide is only the first step; the real challenge is deciding which candidates are worth paying to test. With experimental validation reaching up to $500 per sequence, hit ranking matters.
Protein-peptide docking tools like HADDOCK3[1] are widely used for screening promising candidates based on binding affinity. While useful, docking relies on a static snapshot of an interaction that is, in reality, highly dynamic.
So we wanted to know: can adding all-atom molecular dynamics (MD) simulations recover more true positives and make peptide screening more cost-efficient? To find out, we benchmarked 47 protein-peptide complexes with experimentally validated affinity measurements (Kd and IC50), then compared HADDOCK rankings against MD-derived rankings.
Need help with your peptide campaign?
We help teams prioritize peptide hits with short MD rescoring. Ask us about collaborations and pilot projects.
Let's Discuss Your ProjectBenchmark Results
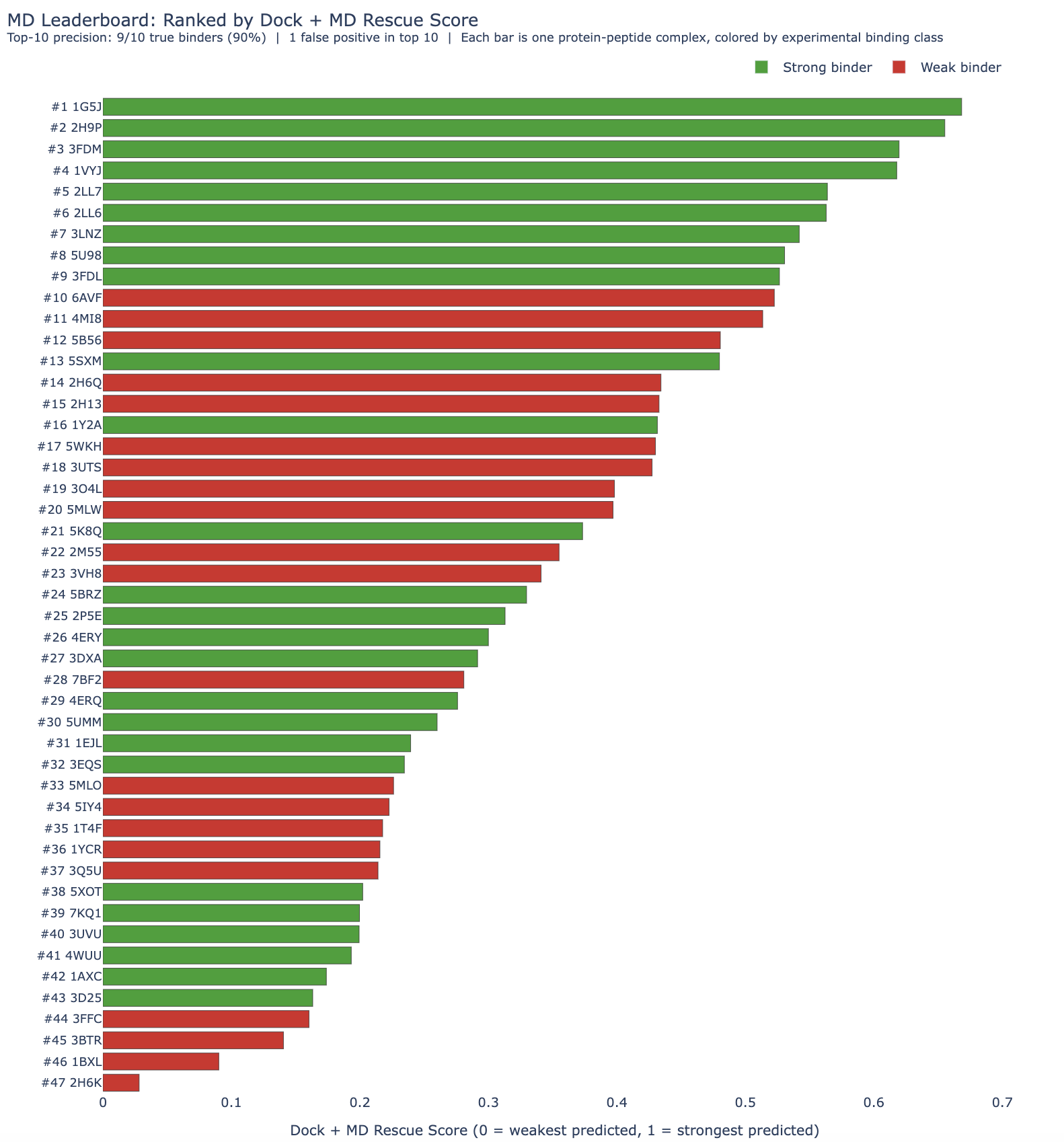
The answer to the question, "Can MD rescoring decrease the number of false positives from protein-peptide docking?" is: Yes. It recovers experimentally validated binders among the top hits that docking missed.
We evaluated 47 protein-peptide complexes with experimentally validated binding affinity data, containing 26 strong binders and 21 weak binders.
We then compared two ranking methods:
1) Docking with HADDOCK3
2) Docking + MD-based rescoring using a composite score comprising mean hydrophobic contacts, average hydrogen-bond contacts, and a shape-complementarity score
To evaluate performance, we counted how many true strong binders were in the top-K, where K represents the number of hits that one would want to test in the lab.
Docking vs. MD Rescoring Performance
HADDOCK correctly identified 4 of the top-5 ranked complexes as strong binders, giving a precision of 80%. Across the top-10, it recovered 7/10 true binders, corresponding to a precision of 70%. Across the larger top-26 queue, it recovered 15/26 true binders.
Docking + MD rescoring recovered 5/5 true binders, reaching 100% precision in the top-5, and found 9/10 true positives, reaching 90%. In other words, MD added two more true binders in the top-10. MD rescoring did not improve enrichment in the larger top-26 assay queue, where it also found 15/26 binders.
True Binder Recovery
| Method | Top-5 Precision | Top-10 Precision | Top-26 Precision |
|---|---|---|---|
| Docking score | 80% (4/5) | 70% (7/10) | 58% (15/26) |
| Docking + MD rescoring | 100% (5/5) | 90% (9/10) | 58% (15/26) |
What this means for screening campaigns
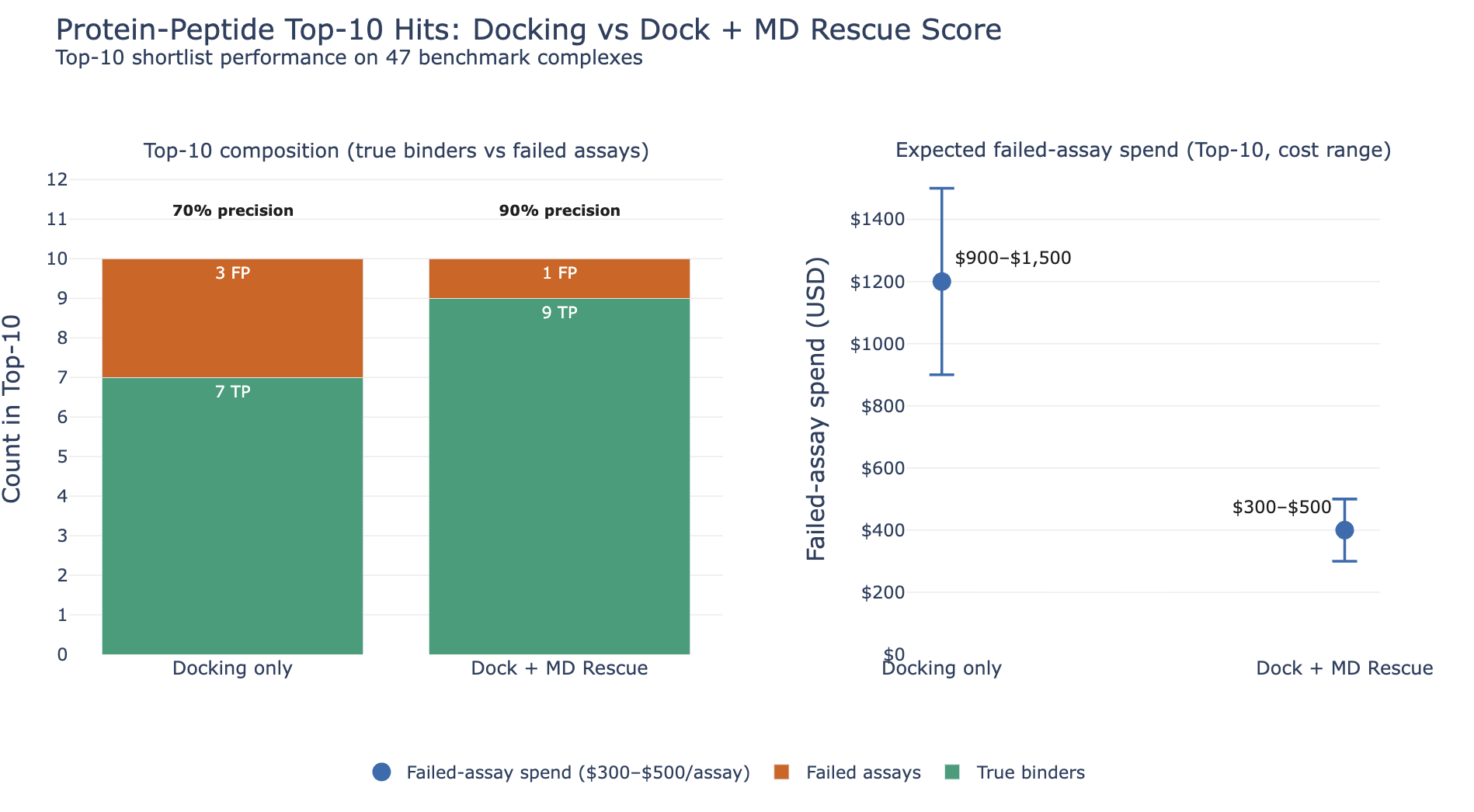
On this 47-complex benchmark, combining HADDOCK3 docking with MD rescoring improved early ranking: it recovered 9 of the top 10 true binders, compared with 7 of 10 for docking alone. That increased top-10 precision from 70% to 90% and avoided two failed assays. Assuming $500 per sequence tested, that corresponds to about $1,000 in wet-lab savings.
MD cost about $1.20-$1.50 per complex for a 10 ns simulation and took roughly 15 minutes of wall time per complex. Across all 47 complexes, that amounts to about $56-$71 in total MD compute cost. After accounting for that expense, avoiding two failed assays corresponds to a net saving of more than $930. In this benchmark, adding MD rescoring delivered a strong return for a very small compute cost.
Our Simple Bio Score measures whether the peptide stays bound in a stable, well-packed interface during the simulation. It combines hydrophobic contacts, hydrogen bonds, and shape complementarity averaged over the trajectory. A caveat to keep in mind is that MD can still reward peptides that are simply sticky, even when they are not truly active. Adding MM/GBSA or MM/PBSA may help reduce those false positives by better accounting for desolvation and interaction energetics.
Because we did not run replicate simulations, these results should be viewed as descriptive, not definitive. They show promising ranking behavior, but a follow-up study with replicates is needed for stronger statistical confidence.
Methods
Benchmark Data Preparation
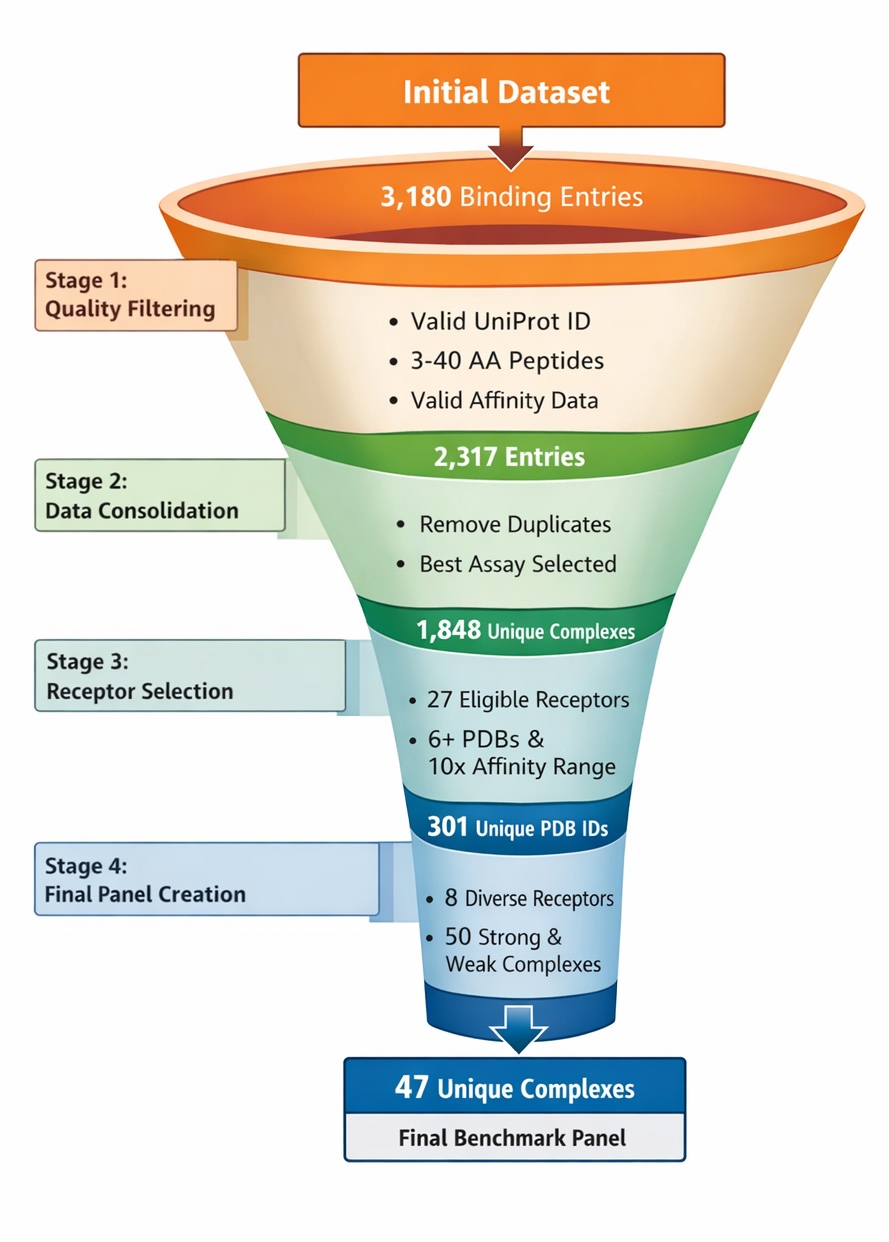
Our goal was to build a benchmark dataset spanning diverse protein receptor families, with each receptor having an equal number of true and false peptide binders supported by experimentally validated binding data. To do this, we curated entries from the Protein-Peptide Interaction Knowledge Base (PPIKB)[2], filtering the original 3,180 experimental binding entries down to a final set of 47 complexes used in this study.
All-Atom Molecular Dynamics Simulations
Molecular dynamics trajectories for all 47 benchmark complexes were generated using the SimAtomic API[5]. Each complex was solvated in explicit TIP3P water. After NVT equilibration, each system was simulated for 10 ns using a 4 fs integration time step with the AMBER ff19SB[6] force field. Each simulation took approximately 15 minutes of wall-clock time to finish, about 1,200 ns/day. Trajectories were analyzed using MDTraj[7]. Frames were subsampled with a stride of 10, and the first 20% of sampled frames were discarded as equilibration. The receptor backbone was aligned to a reference frame, excluding the peptide chain, prior to all spatial analyses. The peptide chain was identified as the smallest protein chain in the topology, and the receptor interface site was auto-detected as the union of receptor residues within 5.0 Å of any peptide heavy atom across the first 3 retained frames.
# MD simulation on 47 complexes with the SimAtomic API. Each complex is packaged as a single ZIP file.
for filepath in ['path/to/complex1.zip', 'path/to/complex2.zip']:
job_id = submit_jobs(config_map, mode="simulation", filename=filepath)Protein-Peptide Docking with HADDOCK3
Protein-peptide docking was performed using HADDOCK3[1] (High Ambiguity Driven protein-protein Docking, version 3). The docking pipeline consisted of five sequential stages: topology generation (topoaa), rigid-body energy minimization (rigidbody), model selection (seletop), semi-flexible refinement (flexref), and final energy minimization in explicit solvent (emref). Docking was run in ab-initio mode with center-of-mass restraints enabled (cmrest = true) and self-contained CNS[8] topology files. For each complex, the larger chain was assigned as the receptor and the smaller chain as the peptide. Sampling parameters were 2 rigid-body models, with the top 1 selected for refinement. The HADDOCK score of the top-ranked model from the emref analysis stage, capri_ss.tsv, was used as the docking score for each complex, where more negative values indicate better predicted binding.
References
- Giulini M, Reys V, Teixeira JMC, Jimenez-Garcia B, Honorato RV, Kravchenko A, Xu X, Versini R, Engel A, Verhoeven S, Bonvin AMJJ. HADDOCK3: A modular and versatile platform for integrative modelling of biomolecular complexes. bioRxiv (2025). doi:10.1101/2025.04.30.651432. This matches the current HADDOCK3 user manual guidance for citing the software.
- PPIKB, Protein-Peptide Interaction Knowledge Base. The official project website describes PPIKB as a data resource of peptide ligand-protein interaction structures and quantitative interaction measurements for peptide drug discovery, and as a structure-activity knowledge base and analysis platform for peptide ligand-protein interactions. Macau Polytechnic University. Accessed April 7, 2026.
- UniProt Consortium. UniProt protein annotation and accession resource. Accessed April 7, 2026.
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Research. 2000;28:235-242. This is the citation recommended by the current RCSB PDB citation policy.
- SimAtomic API documentation. Internal platform documentation used for batch submission and workflow execution in this study.
- Tian C, Kasavajhala K, Belfon KAA, Raguette L, Huang H, Migues AN, Bickel J, Wang Y, Pincay J, Wu Q, Simmerling C. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. Journal of Chemical Theory and Computation. 2020;16:528-552.
- McGibbon RT, Beauchamp KA, Harrigan MP, Klein C, Swails JM, Hernandez CX, Schwantes CR, Wang LP, Lane TJ, Pande VS. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophysical Journal. 2015;109(8):1528-1532.
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR System (CNS): A New Software System for Macromolecular Structure Determination. Acta Crystallographica Section D. 1998;54:905-921. This is the citation requested on the official CNS license page.