Protein + ligand (PDB/mmCIF, SDF)
AI-assisted or traditional
GPU MD presets or custom
Plots, MM/PBSA, export
Built on trusted open-source engines (OpenMM, AMBER force fields, AutoDock Vina)
Advanced molecular simulation — accessible in your browser.
SimAtomic supports key stages of your computational pipeline
Screen compounds with AI-assisted docking, then validate candidates with fast MD and ranking.
Study protein-protein interactions and design better biologics with ensemble-based analysis.
Affordable access to enterprise-grade simulation tools for universities and research institutions.
Credits: 1 credit = 1 min (standard) • ~60 credits/hour
View burn rates
Standard class handles most typical workflows.
Compute Class
Credits/min
Best For
Standard
1
Most MD simulations
High-Memory
1.5
Large systems (>500k atoms)
Multi-GPU
4
Enhanced sampling
100 free credits to run ~1–2 complete simulations. Enough to validate your workflow before committing.
Plan your simulation workload and find the right package
Estimates based on typical protein-ligand systems. Actual usage varies with system complexity and simulation parameters.
Programmatic access to SimAtomic’s simulation and analysis pipeline (included on Pro+ plans).
The SimAtomic API provides programmatic access to our cloud-based molecular dynamics and binding free energy analysis tools. Run simulations, analyze trajectories, and calculate binding energies — through simple REST endpoints.
GPU-accelerated MD
TICA + Clustering
Binding energies
# Initialize the SimAtomic client
from simatomic_client import SimAtomicClient
client = SimAtomicClient(api_key="YOUR_API_KEY")
# Submit a simulation job
job_id = client.run_job(
"complex_input.zip",
config={"md_steps": 500000}
)
# Poll for results
results, _ = client.poll_job(job_id)
if results['job_status'] == "success":
client.download_results(job_id)Access NVIDIA GPUs for accelerated MD without managing infrastructure.
Monitor job status, runtime, and credits consumed per run via the dashboard or API.
MM-PBSA calculations on representative structures for meaningful affinity estimates.
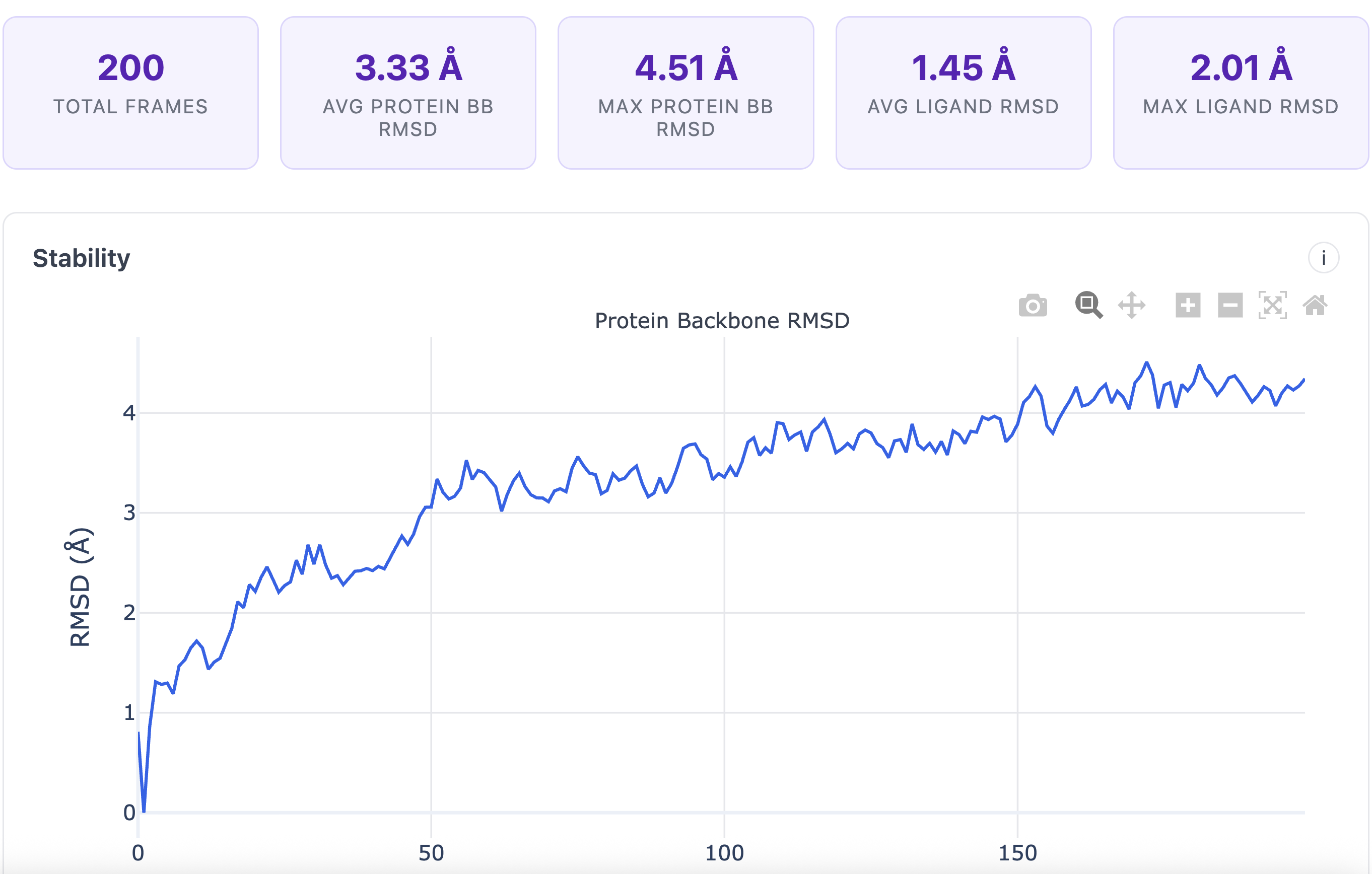
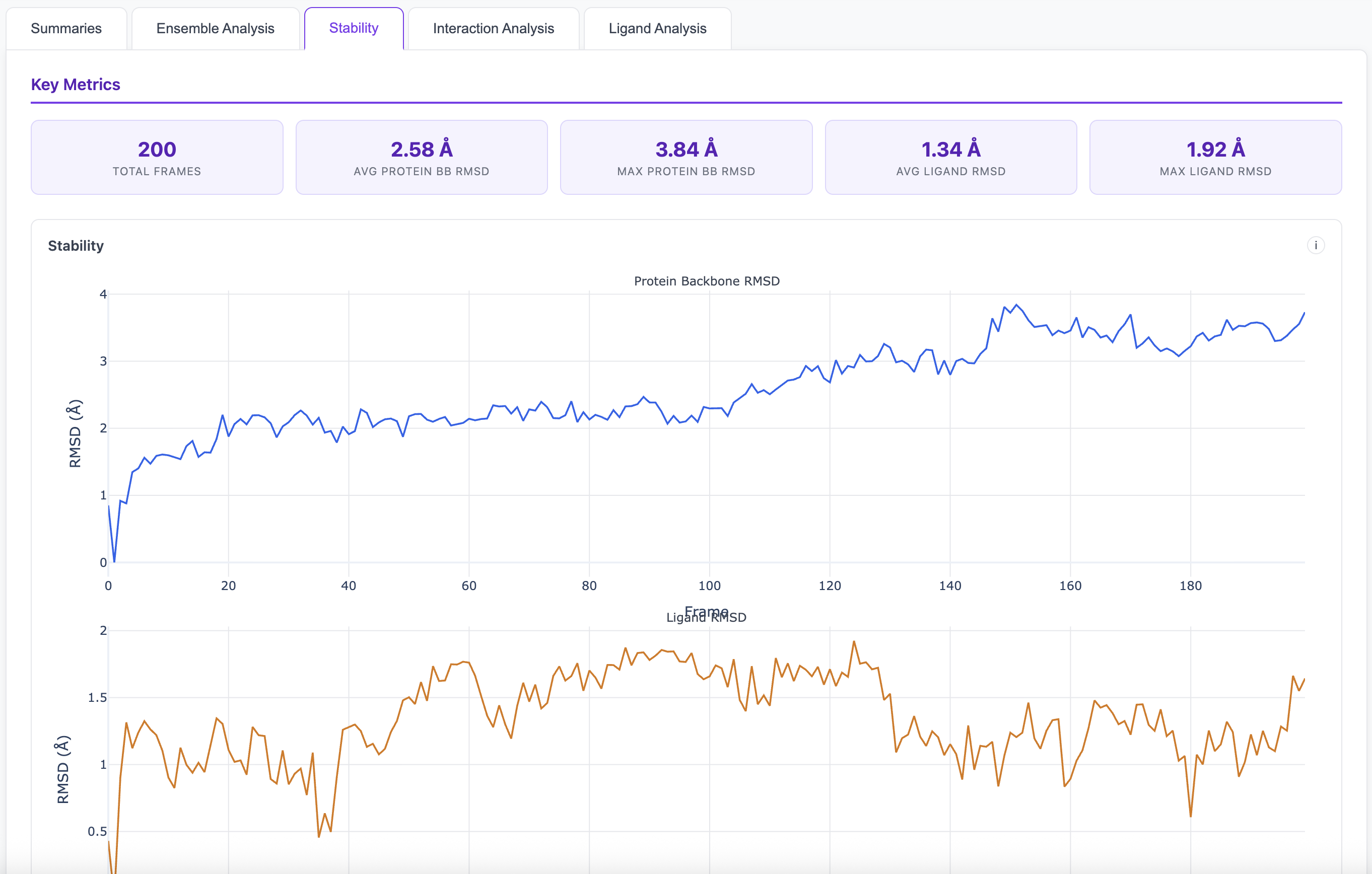
Analyze trajectories with dimensionality reduction and clustering to identify conformational states and stable poses.
SimAtomic supports PDB and SDF files for protein structures. Our platform automatically handles structure preparation, cleaning, protonation and parameterization.
SimAtomic is built on widely used open-source tools: OpenMM for GPU molecular dynamics and AMBER-family force fields for standard biomolecular simulations. Docking can be run with AutoDock Vina (or you can upload docking poses from your existing workflow).
SimAtomic adds the pieces that make this usable for non-specialists: automated setup, validated presets, built-in analysis, and per-job run metadata so results are reproducible.
Tool names are for identification only and do not imply endorsement.
Simulation time depends on system size and desired simulation length. A typical 100ns simulation of a protein-ligand complex completes in a few hours on GPU (varies by parameters).
Data is encrypted in transit (TLS). We're adding additional at-rest protections and can support on-premise deployment for strict governance needs.
Yes, we offer discounts for academic researchers and educational institutions. We are open to both industry and academic collaboration. Contact us to learn more.
Yes. Our REST API and Python client make it easy to integrate SimAtomic into existing workflows. We also support custom integrations for enterprise customers.
All plans include email support with 1-business-day SLA. Pro+ plans include priority queue and escalation handling. Enterprise customers get a dedicated support contact and optional training.
Need something custom? On-prem / VPC · API + pipelines · Training Contact Sales →
Replies within 1 business day. Priority queue + escalation for Pro+ plans.
Every job exports full metadata: parameters, versions, timestamps.
TLS encryption. 30-day retention. On-prem available for Enterprise.
Logos are for identification only and do not imply endorsement.

Ingrid
Barbosa-Farias
Founder
SimAtomic was built by researchers who understand the pain points of computational drug discovery. Built with working scientists in mind—standard protocols, reproducible reports, cost-visible runs.
We built SimAtomic to solve the problems we faced: days wasted on installs and configuration, brittle workflows that break between projects, and opaque costs that make budgeting impossible. The platform delivers production-grade GPU simulations with standard protocols and reproducible reports— accessible to teams of any size.
Join researchers using SimAtomic to streamline molecular simulation.
No credit card required • 14-day free trial • Cancel anytime
Questions or issues? We'll get back to you within 1 business day.