The Problem
Adding a short molecular-dynamics (MD) step after docking in virtual drug screening can cut wet-lab costs by > 50%.
Savings that matter especially for startups and small biotechs needing to stretch their runway, yet few are using it.
Key Benefits of MD Rescoring
- 🔸 <5 ns "shake-out" MD run + MM/PBSA rescoring can more than double confirmed hit-rate by removing docking false-positives (Graves 2008; Brooijmans 2010).
- 🔸 Wet-lab costs scale almost linearly with compounds tested (~$800/compound). Twice the hit rate means half the compounds and half the spending.
- 🔸 A few GPU minutes per ligand cost pennies but can save hundreds or thousands in assays.
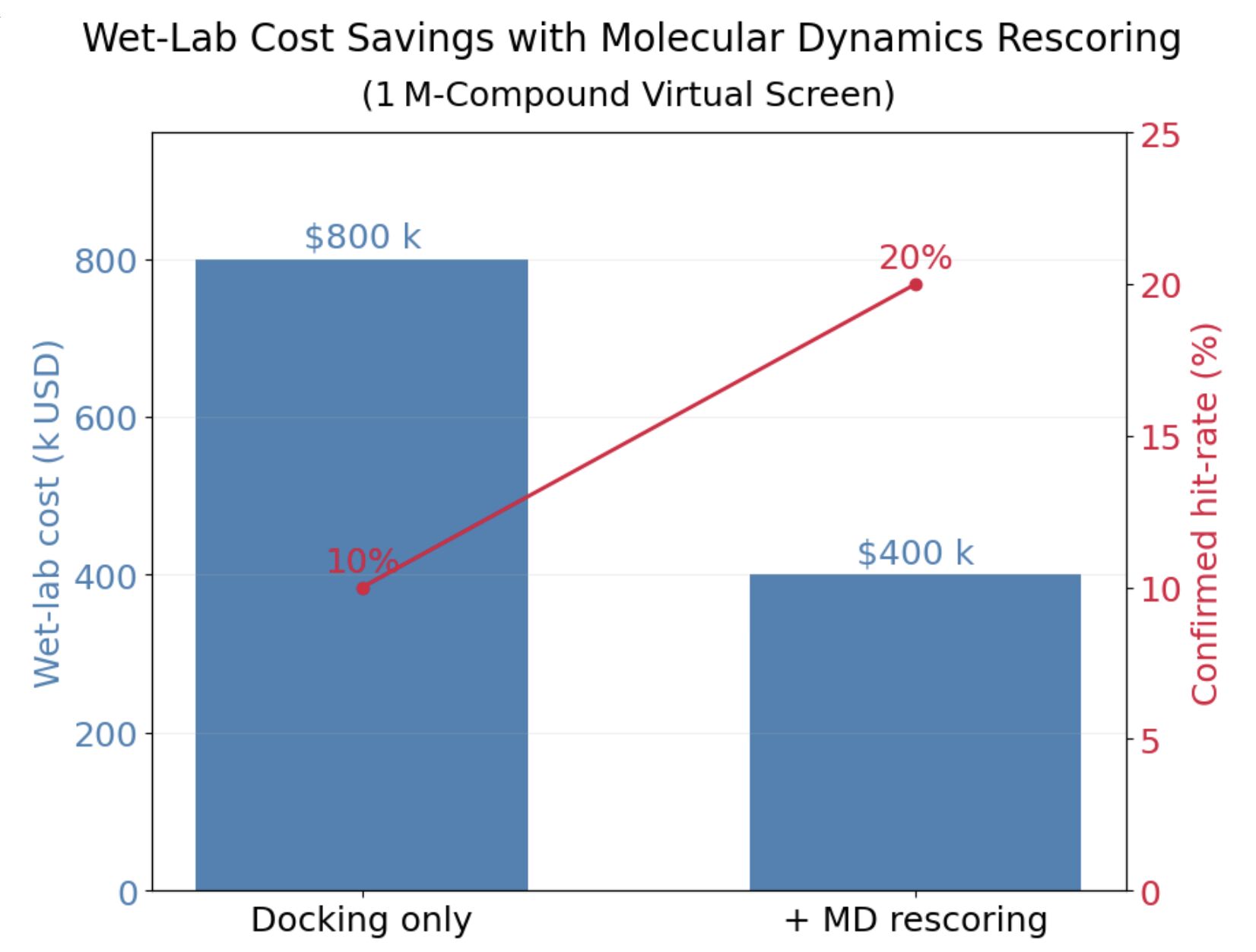
Back-of-the-Envelope Example
Consider a 1M-compound screen:
Docking Only
→ 10% hit rate (100/1,000) ≈ $800k
Docking + MD
→ 20% hit rate (100/500) ≈ $400k
Result: $400k in savings - enough to fund additional research or extend runway significantly.
Why This Works
Traditional docking algorithms often produce false positives due to:
- Rigid receptor assumptions
- Limited sampling of conformational space
- Inadequate treatment of solvent effects
Short MD simulations address these limitations by:
- Allowing protein-ligand complexes to relax
- Sampling multiple conformations
- Providing more accurate binding energy estimates via MM/PBSA
This approach typically requires only 2-5 minutes of GPU time per compound, costing pennies while potentially saving thousands in wet-lab expenses.
Getting Started
Feel free to reach out if you are planning a screening campaign. We're happy to chat about optimizing your virtual screening pipeline for maximum cost efficiency.
Ready to Optimize Your Screening Campaign?
Contact us to discuss how MD rescoring can reduce your wet-lab costs and improve hit rates.
Get in TouchReferences
- Graves, A. P., et al. (2008). "Rescoring docking hit lists for model cavity sites: predictions and experimental testing." Journal of Molecular Biology, 377(1), 317-328.
- Brooijmans, N., & Kuntz, I. D. (2010). "Molecular recognition and docking algorithms." Annual Review of Biophysics and Biomolecular Structure, 32, 335-373.